Autora: Jéssica de Almeida Kulevicz
Colaboradores: Ana Carolina Augusto Silva, Fabiana B. Z. Motizuki e Thalia Macaris
O que é a Coreia de Huntington (DH)
A Coreia ou Doença de Huntington é uma doença hereditária neurodegenerativa progressiva e letal, suas características principais são a tríade: distúrbios de movimento, distúrbios psiquiátricos e demência. A palavra Coréia veio do grego significando “dança”, uma alusão aos movimentos musculares irregulares e involuntários manifestados nos pacientes com a doença.
É uma doença de herança autossômica dominante* com penetrância completa* causada por uma repetição da trinca de nucleotídeos Citosina-Adenina-Guanina (formadores do DNA) presentes no cromossomo 4. A probabilidade dos filhos de uma pessoa afetada apresentarem a doença é de 50%.
Na maioria dos casos o quadro clínico característico aparece entre 30 e 40 anos, já na infância e adolescência os sintomas aparecem em um número menor de casos. Este quadro inicia-se com movimentos involuntários dos músculos sem que ocorra perda da sensibilidade ou consciência, evoluindo gradualmente até a morte.
A incidência da Coreia de Huntington varia de 5 a 10 casos por 100.000 indivíduos e se distribui no mundo todo. Maracaibo e Venezuela apresentam grande incidência no número de casos e para o Brasil não foram encontrados dados reais sobre a doença. A DH independe do gênero, condições socioeconômicas e grupo étnico.
*Herança autossômica dominante: quando o fenótipo (características morfológicas, fisiológicas e comportamentais de um indivíduo) é expresso da mesma maneira em homozigotos e heterozigotos.
*Penetrância completa: o alelo é expresso em 100% dos indivíduos, ou seja, se possuir o alelo mutante irá apresentar a doença.
Histórico

{kind=link}
Modificado de: http://coreiadehuntington.blogspot.com.br/p/fotos.html
A doença foi descrita inicialmente em 1972 pelo médico norte-americano George Huntington que relatou três características da síndrome: A natureza genética, se um ou ambos os pais apresentarem suas manifestações seus filhos provavelmente também sofrerão da doença, porém se estes não possuírem os genes os netos estarão livres; A tendência à insanidade, pois o desenvolvimento da doença altera a mente e leva os enfermos a loucura, podendo levar ao suicídio; Manifesta-se, como doença grave, principalmente na fase adulta, depois dos 40 anos. 1 Em 1877, através de estudos neuropatológicos em cadáveres, foi demonstrado que os sintomas da DH estão associados a alterações em uma região específica do cérebro localizada nos núcleos da base* chamada núcleo caudado. Anos mais tarde, outras pesquisas mostraram que as alterações degenerativas não se restringem apenas a essa região afetando todo o estriado (núcleo caudado e putâmen). Em 1983 o gene relacionado à Coréia de Huntington foi o primeiro responsável por alguma doença a ser localizado em humanos por análise de ligações do DNA e em 1993 foi isolado pelo “Huntington Disease Collaborative Research Group” e descoberto que a causa da doença é o aumento da repetição dos trinúcleotídeos citosina-adenina-guanina (CAG) do geneIT15 no braço curto do cromossomo 4.
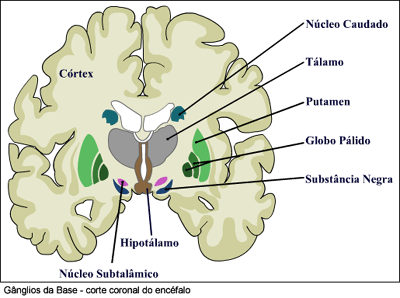
{kind=link}
FIGURA 2 Fonte: http://www.virtual.unifesp.br/unifesp/apoptose /restrito/parkinson_celular1.htm
*Núcleos da Base: São estruturas do cérebro que estão relacionados com o controle da postura e do movimento. Anatomicamente são divididos em núcleo caudado, putâmen e globo pálido e estão funcionalmente muito relacionados entre si. O núcleo caudado e o putâmen juntos formam o estriado(figura 2).
Alterações gênicas
A Coreia de Huntington é uma doença de causa exclusivamente genética e possui herança autossômica dominante de penetrância completa, sendo que todos os indivíduos que possuírem a alteração para DH apresentarão os sintomas.
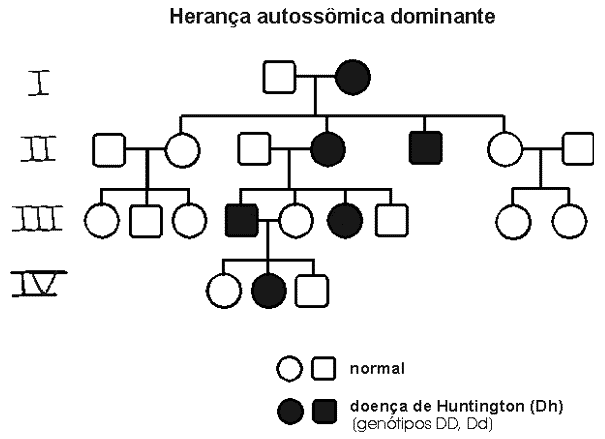
{kind=link}
O gene responsável pela doença é chamado IT15, está localizado no braço curto do cromossomo 4 (4p 16.3) e a mutação ocorre na região 5', com o aumento do número das repetições da trinca de nucleotídeos CAG. Em pessoas normais essa repetição varia de aproximadamente 9 a 34 enquanto na DH o número passa de 40 repetições e quanto maior o número mais precoce é a manifestação da doença, ocorrendo cada vez mais cedo nas próximas gerações, fenômeno chamado de “antecipação”.
Fisiopatologia
O gene IT15 que sofre mutação na DH é responsável por produzir uma proteína chamada huntingtina e o efeito das repetições de CAG é a formação de múltiplos aminoácidos glutamina, maior que o normal, da proteína. Essas cadeias poliglutamínicas se agregam no interior da célula nervosa, levando a uma alteração funcional e morte neuronal. Outra hipótese pode ser que ao se acumular na célula, as poliglutaminas, causam reações anormais de ligação a proteínas e interferem em outros processos celulares.
Essa proteína huntingtina parece ter um importante papel no transporte de vesículas no interior da célula e o RNAm responsável pela síntese da proteína (RNAm Huntington) é encontrado em células de todo o corpo, e está presente em níveis elevados principalmente nos neurônios. Nas células do sistema nervoso, a proteína se encontra no citoplasma neuronal e é vista nos axônios, dendritos e corpo celular.
Tamminga et cols mostraram que a proteína, no citoplasma, teria uma participação nos processos de migração vesicular e exocitose de neurotransmissores e enzimas, além de um certo papel no mecanismo de morte celular através de uma ligação a uma proteína neuronal para formar um complexo tóxico para a célula. Estudos de Reddy el cols demonstraram que enquanto em células normais a huntingtina está presente no citoplasma, nas células mutantes ela aparece no núcleo celular, indicando uma alteração no mecanismo de ação da proteína.
Através de outras pesquisas mostrou-se que a expressão do RNAm Huntington não se altera em pessoas com a doença, pois os locais onde há maior gravidade patológica na DH (núcleo caudado e putâmen) possuem outros fatores que aumentam sua vulnerabilidade à degeneração quando comparados a outras áreas do cérebro
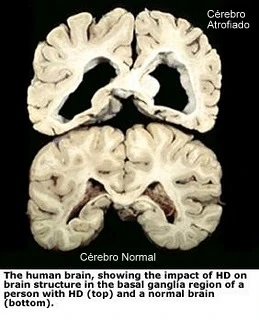

{kind=link}
FIGURA 4 Modificado de: http://lapatufsj.blogspot.com.br /2012/07/doenca-de-huntington.html
Os pacientes portadores do gene mutante apresentam uma atrofia dos núcleos caudados e putâmen e quando a doença está avançada, também uma atrofia no córtex cerebral* (figura 4). Microscopicamente ocorre perda dos corpos celulares de neurônios secretores de GABA e acetilcolina como seus principais neurotransmissores. Os neurotransmissores são substâncias químicas produzidas por esses neurônios e utilizadas para enviar sinais a outras células do corpo. Os neurônios gabaérgicos (produtores do neurotransmissor GABA) inibem o globo pálido e a substância negra e sem essa inibição ocorrem descargas espontâneas de atividade dessas áreas dos núcleos da base cerebrais causando os movimentos característicos da doença. A perda dos neurônios colinérgicos (produtores do neurotransmissor Acetilcolina), principalmente em áreas do córtex cerebral, provavelmente é responsável pela demência. 3
*Córtex cerebral: É a camada que forma a superfície externa do cérebro e ele é responsável por funções como percepção consciente, pensamento, memória e intelecto. 2
Diagnóstico
O diagnóstico da Doença de Huntington é baseado nas manifestações clínicas características e na história familiar. A tomografia computadorizada e a ressonância magnética auxiliam como diagnóstico por imagem e mostram o núcleo caudado e o putâmen atrofiados.
A confirmação da doença é feia através do teste genético. O exame é feito utilizando uma técnica chamada PCR que mostra o número de repetições CAG que o DNA analisado possui. Se as repetições forem maiores que 40 o diagnóstico será positivo para Huntington.
Apesar de ser o exame mais confiável, o exame genético, quando preditivo, envolve algumas questões éticas, jurídicas e discriminatórias. O teste preditivo é um teste para pessoas que estão sob o risco de ter a DH (história familiar positiva, por exemplo), mas ainda não apresentam os sintomas, e apesar de não haver tratamento ou forma de abrandar a progressão da doença, alguns sintomas podem ser controlados inicialmente por medicamentos. Também pode trazer algum benefício emocional ao paciente ao saber que irá apresentar a doença futuramente, assim como malefício, como no caso da descriminação.
Os erros de diagnósticos mais frequentes são três casos: quando o paciente possui um quadro clínico da DH sem a história familiar positiva para a síndrome, em pacientes com risco de desenvolver a DH sem movimentos coreicos (movimentos involuntários e irregulares) e que apresente sintomas psiquiátricos que não sejam consequentes da Coréia de Huntington e pacientes com coréia provocada por outra doença.
O diagnóstico diferencial para essa doença pode ser bastante amplo e doenças como a neuroacantocitose, coréia familiar benigna, atrofia dentatorubropalidoluisiana (DRPLA), doenças priônicas, doença de Machado-Joseph (SCA-3), entre outros distúrbios, apresentam manifestações muito semelhantes a da DH.
Sinais e sintomas – evolução
Os sintomas da Coreia de Huntington são divididos em três categorias: os motores como a coreia que são os movimentos involuntários, distonia (contrações musculares), bradicinésia (lentidão dos movimentos), ataxia (falta de coordenação dos movimentos), dificuldade na fala e na deglutição, entre outros, cognitivos, como perda de memória, envelhecimento mental precoce, perda de concentração e dificuldade na realização de tarefas e emocionais, como a depressão, irritabilidade, ansiedade, estado de desinteresse e mudanças no comportamento.
A DH se manifesta de forma variada em diferentes pessoas, mas apresenta um desenvolvimento em que a doença se agrava com o passar do tempo.
Inicialmente ocorrem poucas mudanças na coordenação e em alguns movimentos involuntários e há dificuldade de pensar sobre problemas. Um sinal da doença é a incapacidade de realizar movimentos sequencias ou movimentos simples repetidas vezes. Nesse estágio o paciente também apresenta humor depressivo ou irritável podendo tratar esses sintomas com medicamentos.
Com o avanço da doença a coreia se agrava, a fala e a deglutição começam a se afetar e as habilidades de pensamento e raciocínio diminuem gradualmente. No estágio mais grave da DH, a coréia pode se intensificar ou, frequentemente, tornar-se rígida. Há também dificuldade de deglutição, desequilíbrio, distonias (contrações musculares involuntárias), ataxia (falta de coordenação nos movimentos), ansiedade ou irritabilidade, delírios e alucinações, podendo evoluir para um quadro de demência além de crises convulsivas e tremores que podem aparecer no curso da doença.
A Coreia de Huntington evolui por 15 a 20 anos após seu início levando e os pacientes morrem, não pela própria doença, mas sim das complicações de imobilidade causadas por ela.
Como tratar
Não há tratamento para curar ou como interromper a progressão da doença. O tratamento dos sintomas comportamentais é feito com antidepressivos e antipsicóticos tradicionais e os sintomas motores com bloqueadores dopaminérgicos.
Referências Bibliográficas
1. CHEMALE F.A., BASSOLS G.F., FERREIRA M.T., ROCHA R.S., ANTONELLO J. Doença de Huntington. Disponível em: <http://genetica.ufcspa.edu.br/seminarios%20textos/Huntignton.pdf> ; Acessado em: 15/11/2012.
2. CROSSMAN A.R.; NEARY D. Neuroanatomia Ilustrada. 4ed. Rio de Janeiro: Elsevier, 2011. p. 134-135, 143-150.
3. GUYTON, A.C.; HALL, J.E. Tratado de fisiologia médica. 12ed. Rio de Janeiro: Elsevier, 2011. p. 733.
4. NUSSBAUM, R.L.; MCINNES, R.R.; WILLARD, H.F. Thompson & Thompson genética médica. 7ed. Rio de Janeiro: Elsevier, 2007. p. 144-145.
5. SILVA E.M.F.,RAMOS J.J.P, GAMA K.J., NASCIMENTO K.S., PIRES L.L., JESUS L.B.B., CARMO M.D., LOBATO N.F., DOMINGUES R. Doença de Huntington: Uma abordagem das características clínicas e fisiopatológicas. Disponível em: <http://www.aprendemos.com.br/doenca-de-huntington/> ; Acessado em: 15/11/2012 .
Links relacionados
1. Associação Brasil Huntington. Disponível em: <http://www.abh.org.br/> ; Acessado em 20/11/2012
2. Doenças Neurodegenerativas. Disponível em:
<http://pt-pr.infomedica.wikia.com/wiki/Doen%C3%A7as_Neurodegenerativas> ; Acessado em 15/11/2012
3. Instituto Virtual de Doenças Neurodegenerativas do Estado do Rio de Janeiro. Disponível em: <http://www.ivdn.ufrj.br/kb_ivdn_v01_00_index.htm> ; Acessado em 20/11/2012
4. Revista Veja. O testamento dentro de cada um. Disponível em: <http://veja.abril.com.br/251109/testamento-dentro-cada-um-p-104.shtml> ; Acessado em 20/11/2012
Vídeos Relacionados
1. Documentário - Convivendo com a doença de Huntington. Disponível em: <http://www.youtube.com/watch?v=Dp-K5ocQVbI>
2. Marcha Coreiforme. Disponível em: <http://www.youtube.com/watch?v=mRp0SMN4xwE>